Alanine dipeptide
Alanine dipeptide is small organic molecule where its geometry is almost expressed with two collective variables, \(\phi\) and \(\psi\). \(\phi\) and \(\psi\) each indicates the diheadral angles (torsion) between centered \(C\) atoms and its neighboring two atoms respectively.
Here, we will going to find multiple pathways of the alaine dipeptide. We used ab initio package VASP, one can use other calculating using slight modification on calc. Coordinates system of the alanine dipeptide is
[-81.89, 71 -> 71.05, -59.29]
C70 : phi = -8.537736562175269e-07 psi = 8.537736462515939e-07
C7eq : phi = -81.89 psi = 74.0
C7ax : phi = 71.05 psi = -59.29
[1]:
import numpy as np
from taps.coords import Cartesian
N = 300
phi = np.linspace(-81.89, 71.05, N)
psi = np.linspace( 74.05, -59.3, N)
coords = Cartesian(coords=np.array([phi, psi])* np.pi / 180, epoch=6, unit='deg')
Inverse Projector
We have represented our system with two angles but to calculate potential energy surface (PES), it is essential to recovor original atomic geometry from collective variables. That is we need, for a given \(\theta\) and \(\psi\), we need a map that sends two coordinate into 22 atoms. Coordinates where we can calculate physical properties of it. Process of inverse projection is tedious, this prepared a pre-scripted code that exactly does that. This script generate the C70 coordinates which represents the \(\phi\) and \(\psi\) (0., 0.) and rotate the atoms to match to that angles.
[2]:
from taps.projectors.alaninedipeptide import AlaninedipeptideInverse
prj = AlaninedipeptideInverse()
print("Before: ", coords.shape)
print("After : ", prj.x(coords).shape)
Before: (2, 300)
After : (3, 22, 300)
Trajectory writing
Someone might want to check this code is indeed working properly, one who want that, use the code below.
from ase.atoms import Atoms
image = Atoms('HCHHCONHCHCHHHCONHCHHH', cell=[15,15,15], pbc=True)
new_coords = prj.x(coords)
filename="test.traj"
from ase.io.trajectory import TrajectoryWriter
trj = TrajectoryWriter(filename, mode="a")
N = new_coords.shape[-1]
#image = self.image.copy()
for n in range(N):
positions = new_coords[:, :, n].T
image.positions = positions
trj.write(image)
VASP calculator
From the projected coordinates generated above, we are going to optimize the structure while keeping two dihedral angles. Our model will uses ab initio pacakge VASP. Unfortunatley, current implementation on ASE does not fully support all the functions on the VASP. Below codes inherits the original Vasp class in the ASE and modify slightly suits that suits our needs. For example, we are going to fix the dihedral angles which needs ICONST, we will generate that. Additionlly, to avoid
similar calculation, if it finds the OUTCAR that has same coordinates withint 0.1 range, it uses that coordinates.
[3]:
import os
from ase.calculators.vasp import Vasp
from ase.calculators.vasp.vasp import check_atoms
from ase.calculators import calculator
class Myvasp(Vasp):
def calculate(self, atoms=None, properties=('energy',),
system_changes=tuple(calculator.all_changes)):
"""Do a VASP calculation in the specified directory.
This will generate the necessary VASP input files, and then
execute VASP. After execution, the energy, forces. etc. are read
from the VASP output files.
"""
check_atoms(atoms)
self.clear_results()
if atoms is not None:
self.atoms = atoms.copy()
command = self.make_command(self.command)
self.write_input(self.atoms, properties, system_changes)
olddir = os.getcwd()
try:
os.chdir(self.directory)
if os.path.isfile('OUTCAR'):
print(self.prefix, 'exist')
errorcode = None
pass
else:
#with open('ICONST', 'w') as f:
# f.write('T 14 21 15 17 0\nT 21 15 17 22 0')
with open('POSCAR', 'a+') as f:
arr = np.random.normal(0, 0.1, (22, 3))
f.write('\n'.join('\t'.join('%0.3f' %x for x in y) for y in arr))
# Create the text output stream and run VASP
os.chdir(olddir)
with self._txt_outstream() as out:
errorcode = self._run(command=command,
out=out,
directory=self.directory)
finally:
os.chdir(olddir)
if errorcode:
raise CalculationFailed(
'{} in {} returned an error: {:d}'.format(
self.name, self.directory, errorcode))
# Read results from calculation
self.update_atoms(atoms)
self.read_results()
calculator.register_calculator_class('Myvasp', Myvasp)
Atomic model
For the atomic construction, we use Atomic Simulation Environment (ASE) package. With the Atoms class in the ASE, we can create image, instance of Atoms class, as reference atomic properties calculation. All the parameters in the image except the positions will be used in the data generation in the ASE, TAPS. We pre-scripted the Alainedipeptide model class which inherits the ASE, for special naming method of the atomic calculation for naming the results.
[4]:
from ase.atoms import Atoms
image = Atoms('HCHHCONHCHCHHHCONHCHHH', cell=[15,15,15], pbc=True)
calc = Myvasp(prec='normal', algo='fast',
command = 'mpirun -np 40 vasp_gam',
nelmin=4, ncore=4, maxmix=20, ismear=0,
isym=-1, istart=2,
smass=-2,
# ibrion=0, mdalgo=1, andersen_prob=0.05, potim=1, tebeg=0,nsw=1,
ibrion=0, mdalgo=1, andersen_prob=0.05, potim=1, tebeg=0,nsw=500,
ediffg= -0.001, encut=520, ediff=1e-5,
isif=0, xc='pbe', ivdw=11, lcharg=True)
image.set_velocities(np.random.normal(0, 0.1, (22, 3)))
image.calc = calc
[5]:
from taps.paths import Paths
from taps.db import ImageData
from taps.ml import Gaussian
from taps.ml.kernels import PeriodicKernel
from taps.models.ase import AlanineDipeptide
model = AlanineDipeptide(image=image, prj=prj,
set_directory=True, directory='./test/vasp/')
hyperparameters = {'sigma_f': 1, 'sigma_n^f': 1e-4,
'sigma_n^e':1e-4, 'l^2': 1}
hyperparameters_bounds = {'sigma_f': (1e-6, 1e4), 'sigma_n^f': (1e-6, 1e-3),
'sigma_n^e':(1e-5, 1e-3), 'l^2': (1e-4, 4)}
model = Gaussian(real_model=model,
kernel=PeriodicKernel(),
hyperparameters=hyperparameters,
hyperparameters_bounds=hyperparameters_bounds)
directory = "./test/"
imgdb = ImageData("./test/alaine_test.db")
paths = Paths(coords=coords, model=model, imgdb=imgdb)
paths.add_image_data(index=[0, paths.N//2, -1])
Newdata []
Newdata [(array([-1.42925012, 1.29241631]), 1)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2)]
[5]:
{'image': [1, 2, 3]}
Paths construction
[6]:
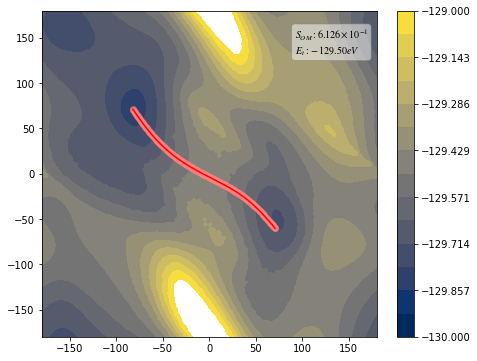
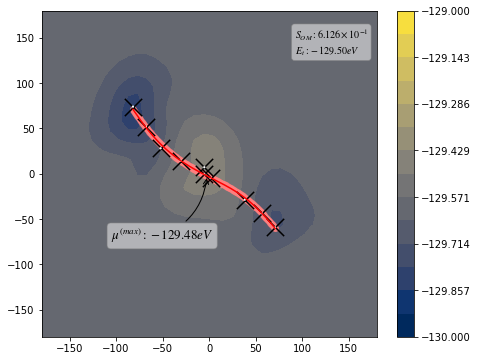
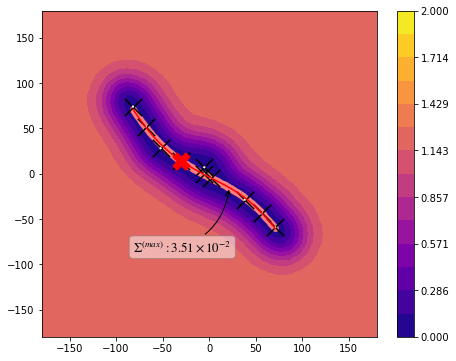
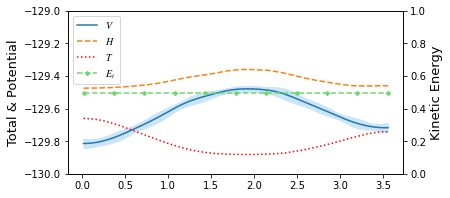
from taps.visualize import view
view(paths, viewer="Alaninedipeptide", gaussian=True)
[6]:
<taps.visualize.view at 0x2ac01b4d2690>
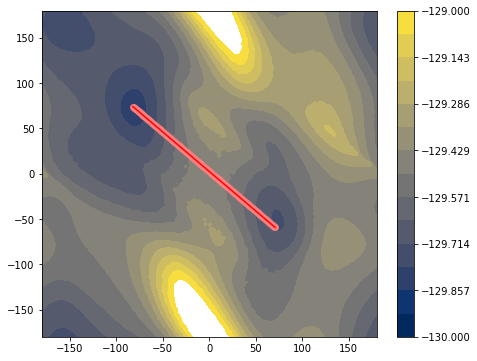
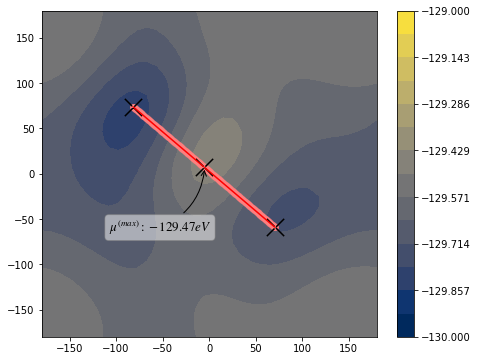
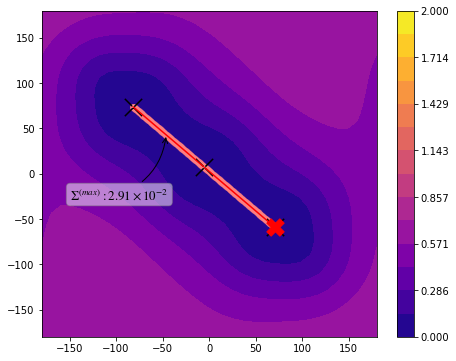
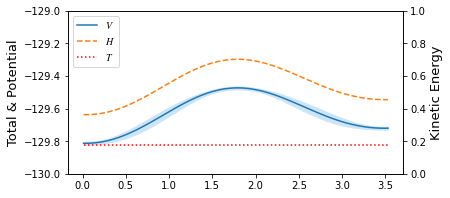
[7]:
paths.model.hyperparameters
[7]:
{'sigma_f': 0.8941067630211492,
'l^2': 1.316186739710582,
'sigma_n^e': 8.875627759142878e-05,
'sigma_n^f': 8.763190535057168e-05}
[8]:
import numpy as np
from taps.coords import Cartesian
N = 300
phi = np.linspace(-81.89, 71.05, N)
psi = np.linspace( 71.05, -60.29, N)
coords = Cartesian(coords=np.array([phi, psi])* np.pi / 180, epoch=6, unit='deg')
[9]:
from taps.paths import Paths
from taps.db import ImageData
from taps.ml import Gaussian
from taps.ml.kernels import PeriodicKernel
from taps.models.ase import AlanineDipeptide
from taps.pathfinder import DAO, GPAO
directory = "./test/"
imgdb = ImageData(directory + "alaine_test.db")
model = AlanineDipeptide(image=image, prj=prj,
set_directory=True, directory='./test/vasp/')
hyperparameters = {'sigma_f': 1, 'sigma_n^f': 1e-4,
'sigma_n^e':1e-4, 'l^2': 1}
hyperparameters_bounds = {'sigma_f': (1e-6, 1e4), 'sigma_n^f': (1e-6, 1e-3),
'sigma_n^e':(1e-5, 1e-3), 'l^2': (1e-4, 4)}
model = Gaussian(real_model=model,
kernel=PeriodicKernel(),
hyperparameters=hyperparameters,
hyperparameters_bounds=hyperparameters_bounds)
finder = DAO(prj_search=False, action_name=["Onsager Machlup", "Energy conservation"],
Et=-129.8, muE=1., gam=1.)
finder = GPAO(real_finder=finder,
label= directory + "gpao_6/1", phases=["auto et"],
plot=view)
paths = Paths(coords=coords, model=model, finder=finder, imgdb=imgdb)
paths.add_image_data(index=[0, paths.N//2, -1])
paths.search()
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3), (array([-0.09013312, -0.00993459]), 4)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3), (array([-0.09013312, -0.00993459]), 4), (array([-0.91083463, 0.48936422]), 5)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3), (array([-0.09013312, -0.00993459]), 4), (array([-0.91083463, 0.48936422]), 5), (array([ 0.66862167, -0.50010278]), 6)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3), (array([-0.09013312, -0.00993459]), 4), (array([-0.91083463, 0.48936422]), 5), (array([ 0.66862167, -0.50010278]), 6), (array([ 0.02986619, -0.08430414]), 7)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3), (array([-0.09013312, -0.00993459]), 4), (array([-0.91083463, 0.48936422]), 5), (array([ 0.66862167, -0.50010278]), 6), (array([ 0.02986619, -0.08430414]), 7), (array([-1.18498414, 0.90364515]), 8)]
Newdata [(array([-1.42925012, 1.29241631]), 1), (array([-0.09013312, 0.12482606]), 2), (array([ 1.24005643, -1.03498025]), 3), (array([-0.09013312, -0.00993459]), 4), (array([-0.91083463, 0.48936422]), 5), (array([ 0.66862167, -0.50010278]), 6), (array([ 0.02986619, -0.08430414]), 7), (array([-1.18498414, 0.90364515]), 8), (array([ 0.99302805, -0.76379324]), 9)]
[10]:
view(paths, viewer="Alaninedipeptide", gaussian=True)
[10]:
<taps.visualize.view at 0x2ac0366db290>